The term Reference Standard Qualifications (RSQ) “is defined as a list of tests, references to analytical procedures, and appropriate acceptance criteria,”1 and incorporate these quantitative reference standards to do so: purity, potency, identification, impurities content, and generally full characterization.
Identity Testing
There are a range of tests to ensure the identity of the drug product or drug substance. These can be done by a variety of equipment, including LC-MS, 1H NMR, 13C NMR, FTIR (Fourier Transform Infrared Spectroscopy), TGA (Thermal Gravimetric Analysis)/DSC (Differential Scanning Calorimetry), and UV-Vis. Appearance testing can be achieved by chiral or ion chromatography, elemental analysis (identifying C, H, and N), TGA/DSC, and XRPD.
Quantitative Testing For quantitative reference standards, there is a standard formula that should utilized (HPLC Purity) x (100% − % Residual Solvents − % Inorganic Impurities)= The Determination and Confirmation of the Assay Value % (i.e., weight %). These component values can be quantified by using HPLC for the purity analysis, residual solvents by LOD or KF Water Content and HSGC, and inorganic impurities by ROI (Residue on Ignition), ICP-MS/OES ((Inductively Coupled Plasma Mass Spectrometry or Inductively Coupled Plasma Optical Emission Spectroscopy). (For a limited number of sample quantities of both residual solvent or inorganic impurities, they can alternatively be tested by TGA.) Finally, to access the weight percentage confirmation, titration and qNMR are excellent techniques.
Concurrent with identity and quantitative testing, storage conditions and stability should be established. This includes storage (container/closure), expiration dating, re-testing procedures, and usage and tracking.
We Can Assist with your Reference Standard Qualifications Needs
SK pharmteco has years of experience and numerous experts in Reference Standard Qualifications. Compendia methods (USP/EP/BP/JP) are utilized in tandem with analytical method development, validation, and transfer to ensure optimal success with your drug substances and drug products. Please contact us with any specific questions or to receive a quote for your RSQ needs.
An Introduction to Dissolution Testing and Development
Dissolution testing is the monitoring of drug substances in a controlled environment from a solid dosage form (i.e., capsules, tablets) to a solution state. These “tests to characterize the dissolution behavior of the dosage form, …also take disintegration characteristics into consideration, are usually conducted using methods and apparatus that have been standardized virtually worldwide over the past decade or so, as part of the ongoing effort to harmonize pharmaceutical manufacturing and quality control on a global basis.”1
Devising a Strategy When devising a dissolution testing strategy, “a simple but broadly applicable analytical method is always desired.”2 Dissolution analysis is generally performed via UHPLC for faster sample analysis due to thenumber of samples required. Creating an analytical method should incorporate guidelines from The European Medicines Agency’s (EMA) International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH), which are also interchangeable with the FDA. These are designed to “avoid redundant testing by (the) industry.”3
It’sthe Media… Dissolution testing is designed to mimic conditions found in the human stomach. As these conditions vary widely from patient to patient, so too should the testing environment. The ranges for the media should allow for various pH levels. These sets should be notated and designed to simulate FaSSGF (fasted state simulated gastric fluid), FeSSIF (fed state simulated intestinal fluid), and FaSSIF (fasted state simulated intestinal fluid). Compendial media is generally HCl or sometimes acetate or phosphate pharmacopeial buffers. As mentioned, with this many simulated environments, along with multiple time sets/points, there will be many samples necessary, and analysis by UHPLC is optimal for timely turnarounds.
We Can Assist with your Dissolution Testing and Development Needs
AMPAC Analytical has years of experience and numerous experts in dissolution testing. We can support release testing, stability, method development, and assist with formulation development through dissolution analysis. We utilize paddle and basket apparatuses in the dissolution phase of testing.Additionally, our equipment offerings include Agilent Infinity II and Thermo Fisher Vanquish Horizon UHPLCs. Please contact us with any specific questions or to receive a quote for your drug product dissolution testing and developmentneeds.
A Brief History of Gas Chromatography Gas chromatography (GC) is one of the most important and prevalent analytical tools available to chemists. It was invented in 1952 by A.T. James and A.J.P. Martin as an outgrowth of research dating back to the previous decade.1-6 Their early techniques on adsorption and partition enabled some of the later developments that A.J.P. Martin spoke about at the 1957 Lansing Symposium, entitled, “Past, Present and Future of Gas Chromatography.” He concluded his address with the following prediction: “If we tie the gas chromatograph to other pieces of laboratory equipment, we have the possibility of almost the automatic chemist…”2 While the idea of an “automatic chemist” hasn’t quite come to fruition, Martin’s belief “that the uniting instrument of the gas chromatograph in the center” of his lab of the future certainly has.2 With the meteoric rise of GC7, its adoption and adaptation continued unabated in the ensuing decades. Then, “The introduction of robust, efficient, and reproducible fused-silica capillary columns and the provision of relatively inexpensive but reliable equipment for GC-MS provided a crucial new impetus in the 1980s.”1 Interestingly, the GC column saw some major advancements within just a few miles of AMPAC Analytical Laboratories. At that location, Walter Jennings and the company he co-founded, J&W Scientific, were instrumental in the development and manufacturing of capillary GC columns. The company was eventually purchased by Agilent in 2001.8,9
GC Capabilities Some of the analyses that GC can do include the separation of compounds in mixtures based on the polarity of the compounds, testing for purity – or for impurities, e.g., and detection of residual solvents. Conversely, with a technique known as preparative chromatography, GC can be used to prepare pure compounds from a mixture. In pharmaceutical analysis, there are additional applications:
Analysis of various functional groups.
Determining purity of pharmaceutical compounds.
Analysis of drugs that are commonly abused.
Determination in pharmaceutical R & D the identity of natural products that containcomplex mixtures of similar compounds.
Use in metabolomics studies.10
GC for Testing Residual Solvents The testing of residual solvents is necessary to ensure potency and, with some solvents, to determine their potential negative effects. GC is an excellent choice to do this. Residual solvents are separated into three classifications by the ICH (International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use):
Class 1 solvents: Solvents to be avoided – Known human carcinogens, strongly suspected human carcinogens and environmental hazards. The Permissible Daily Exposure (PDE) of these solvents used in pharmaceutical products can range from 2 to 1500 parts per million (PPM).
Class 2 solvents: Solvents to be limited – Non-genotoxic animal carcinogens or possible causative agents of other irreversible toxicity such as neurotoxicity or teratogenicity and solvents suspected of other significant but reversible toxicities. The PDE for these solvents ranges from 50 to 3880 ppm.
Class 3 solvents: Solvents with low toxic potential – low toxic potential to man requiring no health-based exposure limit. Class 3 solvents have PDEs of 50 mg or more per day.11
Within these three classifications, some of the most commonly used solvents are listed:
Benzene (class 1)
Acetonitrile, Cyclohexane, Hexane, and Methanol (class 2)
Acetic Acid, Acetone, and Heptane (class 3).11,12
Headspace GC (HSGC) and Direct Injection GC In addition to the numerous GC developments such as column type, phase and coating techniques, and multidimensional GC, two types of sampling methods arose: direct injection (“DI”) and headspace (“HSGC”). In the former, the sample is injected “directly” into a typical sample injector of the GC column. In HSGC, when heated, “the more volatile compounds will tend to move into the gas phase (or headspace) sample. The more volatile the compound, the more concentrated it will be in the headspace. Conversely, the less volatile (and more GC-unfriendly) components that represent the bulk of the sample will tend to remain in the liquid phase.“13
Therefore, by extracting the “headspace vapor and injecting it into a gas chromatograph, there will be far less of the less-volatile material entering the GC column, making the chromatography much cleaner, easier, and faster.”13 Generally, HSGS is much cleaner and results in less wear on the column. For more on comparative techniques with DI and HSGC, please see the first entry in the “Resources” section below.
We Can Assist with your GC Needs
AMPAC Analytical has years of experience and numerous experts in Gas Chromatography who can assist with method development for analyses for both common and atypical sample matrices that will allow you to stay ahead of evolving regulatory concerns. Please contact us with any specific questions or to receive a quote for your GC needs.
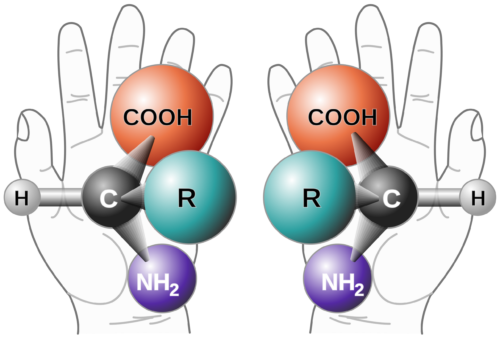
Background on Chiral Purity Chirality refers to the phenomenon that occurs when a mirror image cannot be superimposed. It is sometimes called “optical rotation”.
The origin is from the late 19th-century Greek word kheir (‘hand’) and is one of the easiest demonstrations of the concept. Although a person’s hands may appear virtually identical, if they were switched, the outcome would be very different. Amino acids and sugars are the chiral building blocks of larger molecules such as peptides, proteins, and nucleic acids. Therefore, those polymers, in turn, are chiral as well.1Molecules with chiral centers may have a different therapeutic impact, and this guides the need to test and control chiral purity. The effects of chiral impurities can result in horrific outcomes, as evidenced by the infamous birth defects associated with Thalidomide2 or as benign as Aspartame and sugars (the D/L sugars) that, when superimposed, can create different taste sensations (sweet versus sour, etc.) or metabolic activity. Each chiral center can generate two enantiomers. “Enantiomers or optical isomers are chiral molecules which are non-superimposable mirror images of each other,”3 and multiple chiral centers can generate diastereomers (non-mirror image isomers).
Determining Purity HPLC has been the primary technique for determining chiral purity, with gas chromatography used occasionally. Measurement of optical rotation is a legacy technique that is fast but not as accurate. Historically, HPLC methods used under normal phase conditions could limit the type of molecules that could be analyzed. However, now modern chiral columns are compatible with reverse phase conditions.
Simulated Moving Bed (SMB) Chromatography as an Option for Chiral Purity Analysis Do you have a partner for chiral separations? AMPAC Analytical has the expertise in performing chiral purity testing, along with the equipment and techniques. Additionally, our parent company, AMPAC Fine Chemicals, has decades of experience conducting chromatographic separations at a commercial scale in a highly regulated environment. Our services include SMB screening, method development, proof-of-concept demonstration, and production. We operate the largest CGMP Simulated Moving Bed (SMB) chromatography unit in the United States. These technologies and expertise are part of a one-stop shop (from 10-millimeter columns up to 1000mm). Our SMB processes can be developed in a few weeks and are easily scalable. In many cases, scale-up from gram to multi-ton quantities can be achieved in fewer than six months. Our facilities include kilo-scale and pilot-scale units to support smaller quantities, also under CGMP conditions. The SMB facilities have been inspected and approved by the FDA for the manufacturing of APIs. AFC has registered four products with regulatory authorities (FDA/EMA) using SMB technology. Along with chiral separations, we can also perform the separation of diastereomers & regioisomers.
Contact us today for information on how we can assist with your raw material, amino acid, drug product, and API chiral purity testing or to learn more about our SMB processes.
The linkage between nitrosamines and cancer was first postulated by William Lijinsky in 1970. Then, in 2018, N-nitroso-dimethylamine (NDMA)) was detected in an active pharmaceutical ingredient, Valsartan (an Angiotensin-II-receptor antagonist). Finally, the FDA issued a guidance for the industry, “Control of Nitrosamine Impurities in Human Drugs”, in the fall of 2020. However, the guidelines continue to evolve. There has been an update in March of 2021, with ongoing risk assessments. Other regulatory agencies have instituted their own, along with updates. For example, since our blog series on nitrosamines, there have been some regulatory updates from the European Medicines Agency (EMA). Their new guidelines are outlined in a document entitled, “Questions and Answers for Marketing Authorization Holders/Applicants on the CHMP Opinion for the Article 5(3) of Regulation (EC) No 726/2004 Referral on Nitrosamine Impurities in Human Medicinal Products.”1 The updated section answers the crucial question, “Which limits apply for nitrosamines in medicinal products?”
The answers are provided with a definition of nitrosamines and acceptable exposures:
“The ICH M7(R1) guideline defines N-nitrosamines as substances of the “cohort of concern” for which limits in medicinal products refer to the so-called substance-specific acceptable intake (AI) (the Threshold of Toxicological Concern, TTC, value of 1.5 ug/day cannot be routinely applied) which is associated with a negligible risk (theoretical excess cancer risk of <1 in 100,000 over a lifetime of exposure). The calculation of AI assumes a lifelong daily administration of the maximum daily dose of the medicinal product and is based on the approach outlined in the ICH M7(R1) guideline as well as the principles described in relation to the toxicological evaluation in the assessment report of the CHMP’s Article 5(3) opinion on nitrosamine impurities in human medicinal products.”1,2 (A previous blog examined TTC, here.)
A Useful List of Nitrosamine Limits
In Appendix 1 found on the EMA site, there is a list of more than eighty nitrosamines listed, along with CAS numbers, known medicinal sources, CPCA (Carcinogenic Potency Categorization Approach) categories, and their guidance publication dates.3,4
Some Caveats
There are some exceptions that should be considered. For example, the EMA states, “The ‘less than lifetime’ (LTL) approach should not be applied in calculating the limits as described above but can only be considered after consultation with competent authorities as a temporary measure until further measures can be implemented to reduce the contaminant at or below the limits defined above.”1
Additionally, those medications intended for advanced cancers also have some exceptions. For example, “If the active substance itself is mutagenic or clastogenic at therapeutic concentrations, N-nitrosamine impurities should be controlled at limits for non-mutagenic impurities according to ICH M7(R1).”
There is also guidance when one or more than one nitrosamines may be present. For the latter, the guidance advises one of two approaches:
The total daily intake of all identified N-nitrosamines is not to exceed the AI of the most potent N-nitrosamine identified.
The total risk level calculated for all identified N-nitrosamines is not to exceed 1 in 100,000. The approach chosen needs to be duly justified by the MAH (Marketing Authorization Holder)/Applicant.1
Final Thoughts on Nitrosamines
Nitrosamine guidance worldwide is ever-evolving, yet the impetus to quantify and regulate them is clear. There will doubtlessly be further updates to regulations. AMPAC Analytical Laboratories – an SK pharmteco company (AAL) is an industry leader in the detection of nitrosamines and other genotoxic impurities (GTI), We have the specialized expertise, equipment, and methodologies to detect these impurities by gas chromatography or high-performance liquid chromatography coupled with mass spectrometry to support your API project. Also, importantly, AAL can assist in navigating those projects within today’s regulatory landscape. Please contact us with any specific questions or to receive a quote for nitrosamines or other GTIs.
Dynamic Vapor Sorption (DVS) is a gravimetric technique used to measure the change in mass of a material in response to changes to surrounding conditions such as temperature or humidity. DVS is primarily used with water vapor but can be applied to other organic solvents as well for the physicochemical characterization of solids.
DVS was developed by Daryl Williams, the founder of Surface Measurement Systems Ltd., in 1991. The company then delivered the first working DVS instrument to Pfizer in 1992.1 Since then, many other equipment manufacturers have entered the field.
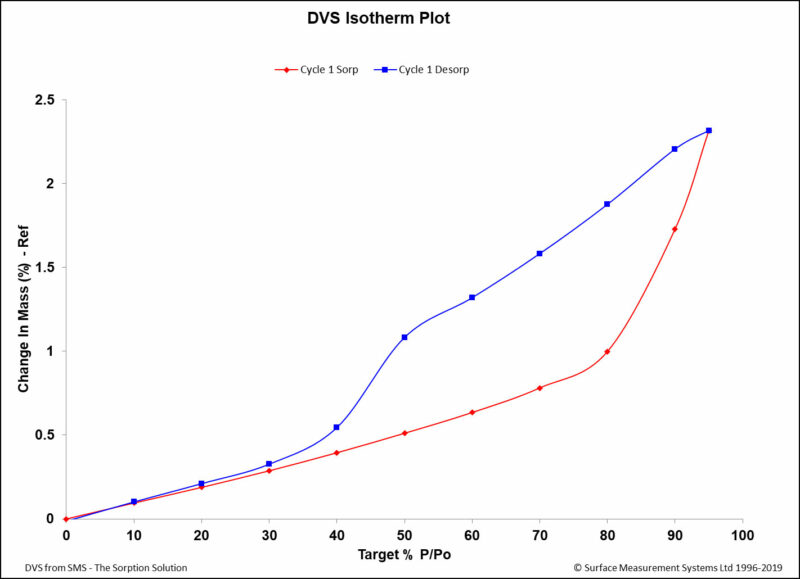
Figure 1: A DVS isotherm plot indicating sorption and desorption rates and hysteresis. The isotherm shows a typical hysteresis curve where the adsorption phase is almost identical to the desorption phase (i.e. reversible). Note that at 80 %RH, there is net sorption of 0.9% between adsorption and desorption traces. The material appears to be slightly hygroscopic according to the definition in Ph. Eur.2
Sorption, Desorption, Absorption, and Adsorption There are five main physical processes that occur during the DVS experiment. The first, sorption, is when a material takes on moisture due to increased humidity. Conversely, desorption is the process that occurs when the material loses moisture due to decreasing humidity. Sorption can be classified as one of two types. Adsorption is moisture that is observed on a surface of a material, while absorption is moisture that has penetrated the surface of a material. The fifth term and the term that relates sorption and desorption is called hysteresis. The overall chart that includes and tracks the sorption and desorption rates and hysteresis is called the isotherm. These curves are crucial for understanding the physicochemical characteristics of a solid, such as porosity, polymorphic change, or liquefying of a sample.2
Applications
There are numerous reasons to utilize DVS, and some of the most common within the active pharmaceutical ingredient (API) industry are:
To determine the sorption isotherm;
To evaluate the hygroscopicity of an API powder;
To compare the hygroscopicity of different solid-state forms: solvates, polymorphs, salts, amorphicity, and cocrystals;
To determine the deliquescence point of a material;
To quantify and qualify the amorphous content in drug substance orexcipient,3 and
To evaluate packaging materials.
Of course, there are a variety of other applications in other industries, including for building materials, food science, cosmetics, coatings, and sealants.
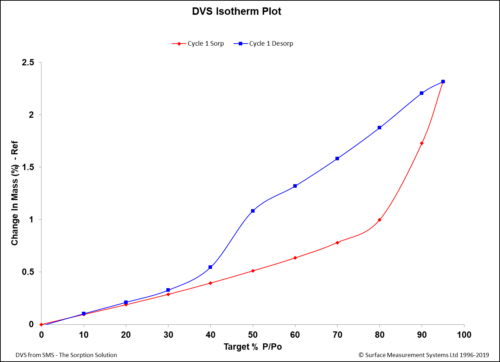
DVS Analysis of APIs For pharmaceutical development, DVS is used for a variety of applications, including screening early drug and excipient candidates, establishing processing parameters, and identifying packaging and storage requirements (Figure 2).4,5
Figure 2: A DVS isotherm of an API showing that the material started to gain significant mass after exposure to relative humidity values of more than 60 %RH. The change here was irreversible, as demonstrated by the desorption curve. DVS could be a useful tool to suggest storage conditions in terms of humidity contents in the surrounding environment.
However, due “to the typically slow establishment of an equilibrium, DVS experiments are rather time-consuming.”4 Nevertheless, “water content of solid active pharmaceutical ingredients and excipients, individually and when formulated in pharmaceutical dosage forms, is a parameter that should be monitored throughout the drug lifecycle.”5
As an analytical technique for APIs, DVS has become a necessary step within drug development and production, reducing issues that can arise during manufacturing, packaging, transportation, solubility, dissolution rate, stability, or storage. AMPAC Analytical’s sister company, SK biotek Ireland Analytical Services, has the experience, technology (including a Surface Measurement System’s DVS Resolution Dual Vapor Gravimetric Sorption Analyser), and support, to assist with this vital testing. Both companies are part of SK pharmteco and can easily transfer your project from either business unit to ensure the most optimal solution and logistical support are provided to meet your product timeline. We invite you to contact our team members and discuss how we can assist with your sorption testing requirements.
Figure 3: A SMS DVS instrument like the one located at SK biotek Ireland.
Effective method development is crucial for the quality control of Active Pharmaceutical Ingredients (API) and Drug Products (DP). Thorough method development enables successful downstream method validation.
The regulatory guidance specifies that:
Method development and validation vary by application (quantitative, qualitative, etc.).
It is phase appropriate.
The client may provide additional guidance/validation criteria.
The validation guidance directs how AMD (analytical method development) is conducted.
Early Adoption of Forced Degradation Analysis It is recommended that forced degradation be performed early in the method development lifecycle and that method parameters are suitable for mass spectrometry. This will prevent many issues that could occur in later stages and ensures the primary purity method is stability-indicating (specificity). When performing forced degradation, these considerations should be weighed:
Always utilize a control sample without exposure to stressors.
The stressors generally consist of acid, base, peroxide, heat, and photolytic conditions. Other stressors may be used based on known material incompatibility
After exposing the compound to these stressors, target 5-20% degradation of the main peak.
If degradation is not observed under reasonable conditions, then the material can be considered stable under those conditions.
Impurity Genesis and Identification Assessment Acceptance criteria should be scaled to impurity levels. How can the method unequivocally assess the analyte of interest in the presence of likely impurities, degradants, and the sample matrix? Additional considerations include:
Is themethod capable of identifying and/or quantifying a specific compound?
Are there solvents present that can interfere with potential impurities?
Known impurities? Are the known impurities stable under the method conditions?
Is the method specific for degradation byproducts for stability-indicating methods?
Cross-Platform Method Robustness Robustness refers to a method’s ability to meet its analytical requirements (system suitability requirements) despite small variations of the method’s parameters, such as discrete changes to a column or sample tray temperature, percent organic modifier, flow rate, and detector wavelength. This capability is typically built into the method during method development.
Precision or Accuracy – You Need Both in Strong AMD AMPAC Analytical development strategy involves the early adoption of forced degradation studies with the goal of every primary purity method being stability-indicating (specificity) and mass spectrometry compatible. The validation strategy is phase-appropriate and application-specific and guides the development strategy. The validation acceptance criteria guidelines are specific to the test methodology, intended use, and level. Finally, method lifecycle performance is assessed, and reevaluation or revalidation can occur.
Contact us today to learn how we can create accurate, precise method development for your API and DP pipeline.
The Cost of Drug Discovery and Development and How to Mitigate It
The path to successful drug discovery and development is extremely long, expensive, and risky and can take between 10 to 15 years at an average cost of more than $1–2 billion for every new drug that is approved for clinical use.1,2 In fact, preclinical drug discovery alone “typically takes five and a half years and accounts for about one-third of the cost of drug development.”3,4 Therefore, even during the earliest stages of a drug product or active pharmaceutical ingredient project, phase-appropriate method development should be instituted to manage costs. This bolsters the chances for success and ensures reliable results, quality management, and reproducibility while avoiding “unreliable results (that) might not only be contested in court but could also lead to unjustified legal consequences for the defendant or to wrong treatment of the patient.”5 At its most basic, phase-appropriate method development maps the “what is needed” to “when it is needed.”6 Effective phase-appropriate method development can provide long-term product support by introducing mass spectrometry compatibility and forced degradation development to ensure your methods are stability-indicating and amenable to unknown impurity identification. By instituting a phase-appropriate method development process, combined with a quality-by-design approacharound each logical sequence of events – and rigorously following it – it is more likely to create a cost-effective, successful outcome as you take the drug product through the regulatory process.
It Can Pay to Outsource
As the incentives for strong phase-appropriate method development increase, so too has the recognition of its value. Unfortunately, “it is not uncommon…to find pharmaceutical companies and contract research organizations (CROs) that are not taking advantage of the phase-appropriate approach and simply reference the typical ICH guidance for analytical items, such as method validation.”7 However, while FDA guidance encourages the use of a phase-appropriate approach, it is lacking in details and requirements. This leaves many companies to seek out ICH guidance as an alternative, conservative approach. Also, within their CGMP quality system, they may find it difficult to accommodate differing levels of CGMP compliance throughout the various clinical phases of development. This is when it might be an opportune moment to consider an outside expert that specializes in phase-appropriate method development processes for drug discovery and validation. A successful yet robust phase-appropriate method development program can balance competing interests and requirements and still provide a regimen that meets the overall development goals without sacrificing any of the requirements of the program.
AMPAC Analytical Laboratories (AAL), an SK pharmteco company, has decades of experience in providing a wide array of release testing services for raw materials, intermediates, APIs, and drug products. Our labs are equipped to handle hazardous, cytotoxic/high potency compounds as well as controlled substances for schedule II through V. Additionally, we have not only the expertise to conduct forced degradation experimentsbut also appropriate instrumentation like mass spectrometers to support later phases of development for your products. Please contact us to discuss how we can ensure the success of your drug discovery and development project and simultaneously reduce risks.






 Gas chromatography (GC) is one of the most important and prevalent analytical tools available to chemists. It was invented in 1952 by A.T. James and A.J.P. Martin as an outgrowth of research dating back to the previous decade.
Gas chromatography (GC) is one of the most important and prevalent analytical tools available to chemists. It was invented in 1952 by A.T. James and A.J.P. Martin as an outgrowth of research dating back to the previous decade.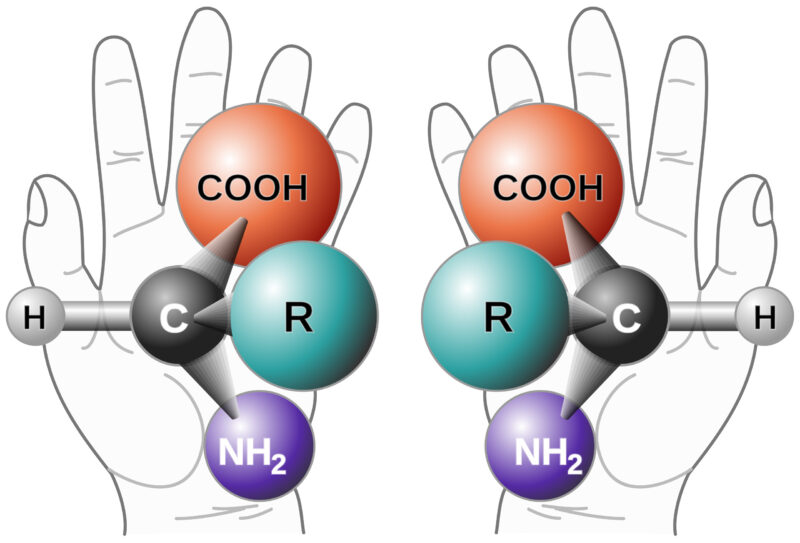
 Background on Chiral Purity
Background on Chiral Purity


 Effective method development is crucial for the quality control of
Effective method development is crucial for the quality control of 

 The path to successful drug discovery and development is extremely long, expensive, and risky and can take between 10 to 15 years at an average cost of more than $1–2 billion for every new drug that is approved for clinical use.
The path to successful drug discovery and development is extremely long, expensive, and risky and can take between 10 to 15 years at an average cost of more than $1–2 billion for every new drug that is approved for clinical use.