
Material science and physical characterization are crucial to ensure drug products’ or drug substances’ identities with measurements and analysis. There are many types of techniques and equipment to do this, each with its own features and qualities. In this blog, the first part of a series, we will review the advantages of a range of techniques and equipment.
 We’ll begin with X-Ray Powder Diffraction (XRPD) or XRD analysis. It is a powerful, non-destructive, and rapid technique for analyzing a wide range of materials (1 µm to 100 mm), including metals, polymers, catalysts, plastics, pharmaceuticals, and other materials. It is used for identification, crystal form characterization, and crystalline content in amorphous products.
We’ll begin with X-Ray Powder Diffraction (XRPD) or XRD analysis. It is a powerful, non-destructive, and rapid technique for analyzing a wide range of materials (1 µm to 100 mm), including metals, polymers, catalysts, plastics, pharmaceuticals, and other materials. It is used for identification, crystal form characterization, and crystalline content in amorphous products.
Inductively Coupled Plasma-Mass Spectrometry (ICP-MS) is useful for the detection of elemental content and elemental impurities. ICP-MS is also

efficient as it is a multi-element technique. Ions are directly detected by an MS detector rather than by emission of light, as in the case of ICP-OES (Inductively Coupled Plasma-Optical Emission Spectroscopy). The ions are separated by a Quadrupole based on the mass-to-charge ratio. The best detection limits are available for most of the elements in ICP-MS as the number of ions produced is high, and although some spectral interference is seen, these are defined and limited.
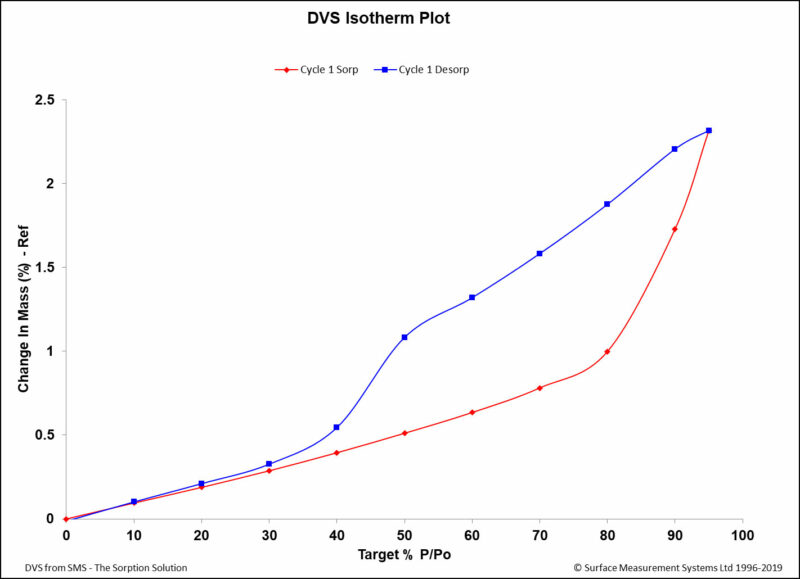
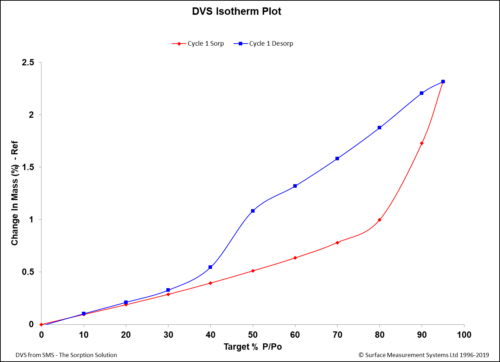
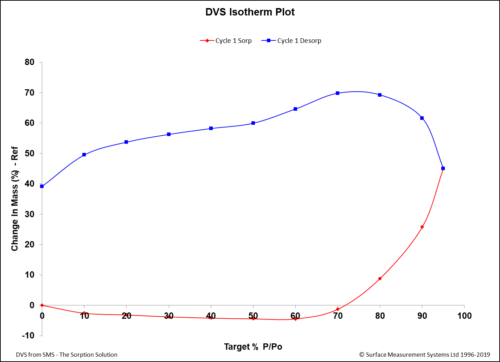
Dynamic Vapor Sorption (DVS) is a gravimetric technique used to measure the change in

mass of a material in response to changes to surrounding conditions such as temperature or humidity. DVS is primarily used with water vapor but can be applied to other organic solvents as well for the physicochemical characterization of solids. Some of the most common uses of DVS include:
- To determine the sorption isotherm;
- To evaluate the hygroscopicity of an API powder;
- To compare the hygroscopicity of different solid-state forms: solvates, polymorphs, salts, amorphicity, and cocrystals;
- To determine the deliquescence point of a material;
- To quantify and qualify the amorphous content in drug substance or excipient, and
- To evaluate the efficacy of packaging materials.
We will wrap up with Differential Scanning Calorimetry (DSC). It is one of the most used thermoanalytical techniques for determining freezing and melting points and phase transitions. Specifically, it measures the heat flow produced by a sample when it is either heated, cooled, or held isothermally at an unchanging temperature. Crystallization behavior and chemical reactions are some other characteristics that can be measured by this technique.
Our next entry will conclude with a look at Thermogravimetric Analysis (TGA), Fourier-Transform Infrared Spectroscopy (FTIR), Nuclear Magnetic Resonance (NMR), and, of course, the definitive analytical technique, Mass Spectroscopy (both GCMS and LCMS).
Resources
- Materials Characterization Techniques – Sam Zhang, Lin Li, Ashok Kumar – Google Books
- https://ampacanalytical.com/laboratory-services/x-ray-diffraction-analysis/
- Dynamic Vapor Sorption
- Setting the Standard with Reference Standard QualificationsRaw Materials Testing: Trust – and Verify – Your Sources
- Elemental Impurities
- DSC Analysis – Fundamentals and Applications (mt.com)







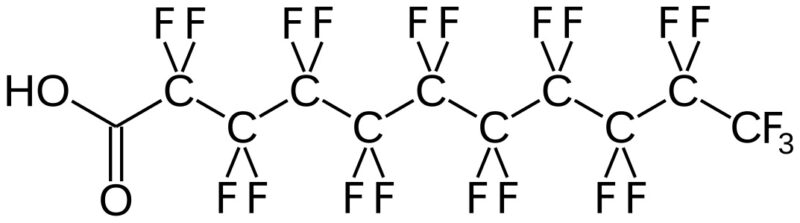
 (Per- and) PolyFluoroAlkyl Substances (PFAS) are a class of ubiquitous chemicals that have been found in water, air, fish, and soil across the nation and worldwide. Known as “Forever Chemicals,” there are thousands of different PFAS, and they are present in consumer, commercial, and industrial products.1 Having one of the strongest bonds in organic chemistry, their structures proved to be resistant to heat, water, oil, and degradation.2 They are found in “food packaging and non-stick cookware, cosmetics, waterproof and stain-proof textiles and carpet, aqueous film forming foam (AFFF) to fight Class B fires, and as part of metal plating processes.”3 Teflon and Scotchgard were two of the pioneering products to utilize these fluoropolymers. The two most common PFAS are perfluorooctanoic acid (PFOA) and perfluorooctane sulfonic acid (PFOS).
(Per- and) PolyFluoroAlkyl Substances (PFAS) are a class of ubiquitous chemicals that have been found in water, air, fish, and soil across the nation and worldwide. Known as “Forever Chemicals,” there are thousands of different PFAS, and they are present in consumer, commercial, and industrial products.1 Having one of the strongest bonds in organic chemistry, their structures proved to be resistant to heat, water, oil, and degradation.2 They are found in “food packaging and non-stick cookware, cosmetics, waterproof and stain-proof textiles and carpet, aqueous film forming foam (AFFF) to fight Class B fires, and as part of metal plating processes.”3 Teflon and Scotchgard were two of the pioneering products to utilize these fluoropolymers. The two most common PFAS are perfluorooctanoic acid (PFOA) and perfluorooctane sulfonic acid (PFOS).
 Gas chromatography (GC) is one of the most important and prevalent analytical tools available to chemists. It was invented in 1952 by A.T. James and A.J.P. Martin as an outgrowth of research dating back to the previous decade.
Gas chromatography (GC) is one of the most important and prevalent analytical tools available to chemists. It was invented in 1952 by A.T. James and A.J.P. Martin as an outgrowth of research dating back to the previous decade.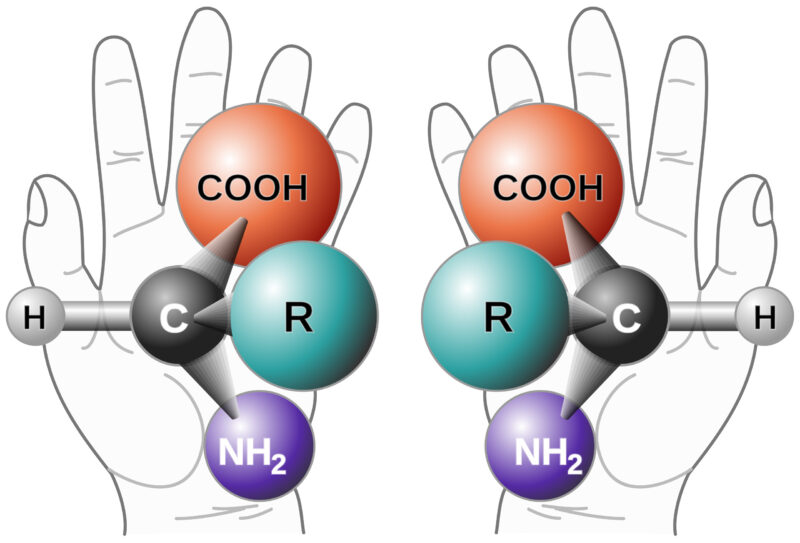
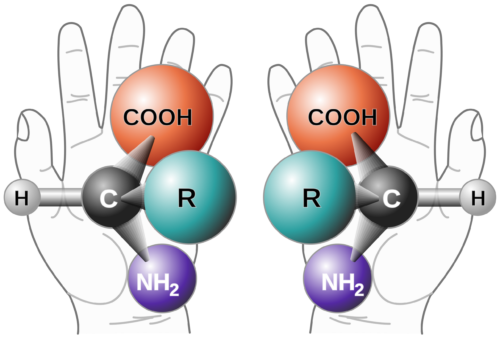 Background on Chiral Purity
Background on Chiral Purity


 Effective method development is crucial for the quality control of
Effective method development is crucial for the quality control of 