The term Reference Standard Qualifications (RSQ) “is defined as a list of tests, references to analytical procedures, and appropriate acceptance criteria,”1 and incorporate these quantitative reference standards to do so: purity, potency, identification, impurities content, and generally full characterization.
Identity Testing
There are a range of tests to ensure the identity of the drug product or drug substance. These can be done by a variety of equipment, including LC-MS, 1H NMR, 13C NMR, FTIR (Fourier Transform Infrared Spectroscopy), TGA (Thermal Gravimetric Analysis)/DSC (Differential Scanning Calorimetry), and UV-Vis. Appearance testing can be achieved by chiral or ion chromatography, elemental analysis (identifying C, H, and N), TGA/DSC, and XRPD.
Quantitative Testing For quantitative reference standards, there is a standard formula that should utilized (HPLC Purity) x (100% − % Residual Solvents − % Inorganic Impurities)= The Determination and Confirmation of the Assay Value % (i.e., weight %). These component values can be quantified by using HPLC for the purity analysis, residual solvents by LOD or KF Water Content and HSGC, and inorganic impurities by ROI (Residue on Ignition), ICP-MS/OES ((Inductively Coupled Plasma Mass Spectrometry or Inductively Coupled Plasma Optical Emission Spectroscopy). (For a limited number of sample quantities of both residual solvent or inorganic impurities, they can alternatively be tested by TGA.) Finally, to access the weight percentage confirmation, titration and qNMR are excellent techniques.
Concurrent with identity and quantitative testing, storage conditions and stability should be established. This includes storage (container/closure), expiration dating, re-testing procedures, and usage and tracking.
We Can Assist with your Reference Standard Qualifications Needs
SK pharmteco has years of experience and numerous experts in Reference Standard Qualifications. Compendia methods (USP/EP/BP/JP) are utilized in tandem with analytical method development, validation, and transfer to ensure optimal success with your drug substances and drug products. Please contact us with any specific questions or to receive a quote for your RSQ needs.
Forced Degradation is the testing of a drug product or drug substance using situations more taxing than those if conditions were simply accelerated. This type of stability testing is designed to demonstrate various degradation pathways within the product or substance and assists with the development of the product itself, along with its packaging. Within forced degradation testing, there is a subset of studies known as photo-stability. The guidelines are outlined by the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) and the FDA. As the ICH Topic Q1B document states, “The intrinsic photostability characteristics of new drug substances and products should be evaluated to demonstrate that, as appropriate, light exposure does not result in unacceptable change.”1
They recommend a range of different studies that address potential areas of concern:
A systematic approach to photostability testing is recommended covering, as appropriate, studies such as:
(i) Tests on the drug substance; (ii) Tests on the exposed drug product outside of the immediate pack; and, if necessary,
(iii) Tests on the drug product in the immediate pack; and, if necessary,
(iv) Tests on the drug product in the marketing pack.1
The type of lights used in photo-stability are also described. They offer options, including:
…any light source that is designed to produce an output similar to the D65/ID65 emission standard…
A cool white fluorescent lamp designed to produce an output similar to that specified in ISO 10977 (1993) …
A near UV fluorescent lamp having a spectral distribution from 320 nm to 400 nm with a maximum energy emission between 350 nm and 370 nm…1
Additionally, temperature control should be maintained to minimize changes and variations.
Finally, these guidelines also provide and elucidate the differences between procedures for drug products and drug substances. It is a crucial reference for administering or simply learning more about photo-stability forced degradation testing.
AMPAC Analytical has the equipment and expertise to provide comprehensive photo-stability studies for your products. Additionally, we have high-resolution and triple-quadrupole mass spectrometers to aid in the identification of potential degradants. Contact us today to learn how AMPAC Analytical can support your photo-stability needs.
Dynamic Vapor Sorption (DVS) is a gravimetric technique used to measure the change in mass of a material in response to changes to surrounding conditions such as temperature or humidity. DVS is primarily used with water vapor but can be applied to other organic solvents as well for the physicochemical characterization of solids.
DVS was developed by Daryl Williams, the founder of Surface Measurement Systems Ltd., in 1991. The company then delivered the first working DVS instrument to Pfizer in 1992.1 Since then, many other equipment manufacturers have entered the field.
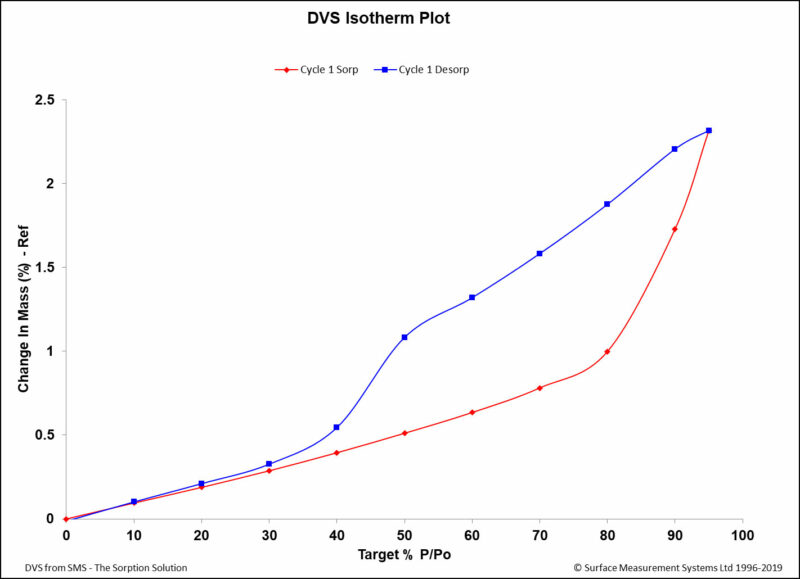
Figure 1: A DVS isotherm plot indicating sorption and desorption rates and hysteresis. The isotherm shows a typical hysteresis curve where the adsorption phase is almost identical to the desorption phase (i.e. reversible). Note that at 80 %RH, there is net sorption of 0.9% between adsorption and desorption traces. The material appears to be slightly hygroscopic according to the definition in Ph. Eur.2
Sorption, Desorption, Absorption, and Adsorption There are five main physical processes that occur during the DVS experiment. The first, sorption, is when a material takes on moisture due to increased humidity. Conversely, desorption is the process that occurs when the material loses moisture due to decreasing humidity. Sorption can be classified as one of two types. Adsorption is moisture that is observed on a surface of a material, while absorption is moisture that has penetrated the surface of a material. The fifth term and the term that relates sorption and desorption is called hysteresis. The overall chart that includes and tracks the sorption and desorption rates and hysteresis is called the isotherm. These curves are crucial for understanding the physicochemical characteristics of a solid, such as porosity, polymorphic change, or liquefying of a sample.2
Applications
There are numerous reasons to utilize DVS, and some of the most common within the active pharmaceutical ingredient (API) industry are:
To determine the sorption isotherm;
To evaluate the hygroscopicity of an API powder;
To compare the hygroscopicity of different solid-state forms: solvates, polymorphs, salts, amorphicity, and cocrystals;
To determine the deliquescence point of a material;
To quantify and qualify the amorphous content in drug substance orexcipient,3 and
To evaluate packaging materials.
Of course, there are a variety of other applications in other industries, including for building materials, food science, cosmetics, coatings, and sealants.
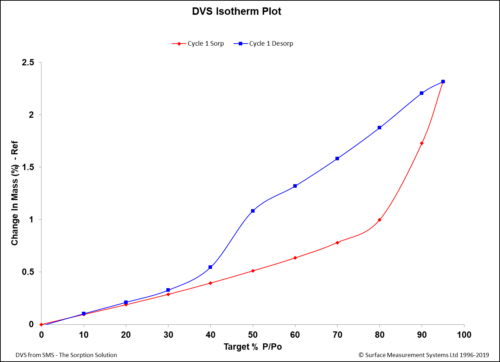
DVS Analysis of APIs For pharmaceutical development, DVS is used for a variety of applications, including screening early drug and excipient candidates, establishing processing parameters, and identifying packaging and storage requirements (Figure 2).4,5
Figure 2: A DVS isotherm of an API showing that the material started to gain significant mass after exposure to relative humidity values of more than 60 %RH. The change here was irreversible, as demonstrated by the desorption curve. DVS could be a useful tool to suggest storage conditions in terms of humidity contents in the surrounding environment.
However, due “to the typically slow establishment of an equilibrium, DVS experiments are rather time-consuming.”4 Nevertheless, “water content of solid active pharmaceutical ingredients and excipients, individually and when formulated in pharmaceutical dosage forms, is a parameter that should be monitored throughout the drug lifecycle.”5
As an analytical technique for APIs, DVS has become a necessary step within drug development and production, reducing issues that can arise during manufacturing, packaging, transportation, solubility, dissolution rate, stability, or storage. AMPAC Analytical’s sister company, SK biotek Ireland Analytical Services, has the experience, technology (including a Surface Measurement System’s DVS Resolution Dual Vapor Gravimetric Sorption Analyser), and support, to assist with this vital testing. Both companies are part of SK pharmteco and can easily transfer your project from either business unit to ensure the most optimal solution and logistical support are provided to meet your product timeline. We invite you to contact our team members and discuss how we can assist with your sorption testing requirements.
Figure 3: A SMS DVS instrument like the one located at SK biotek Ireland.
The Cost of Drug Discovery and Development and How to Mitigate It
The path to successful drug discovery and development is extremely long, expensive, and risky and can take between 10 to 15 years at an average cost of more than $1–2 billion for every new drug that is approved for clinical use.1,2 In fact, preclinical drug discovery alone “typically takes five and a half years and accounts for about one-third of the cost of drug development.”3,4 Therefore, even during the earliest stages of a drug product or active pharmaceutical ingredient project, phase-appropriate method development should be instituted to manage costs. This bolsters the chances for success and ensures reliable results, quality management, and reproducibility while avoiding “unreliable results (that) might not only be contested in court but could also lead to unjustified legal consequences for the defendant or to wrong treatment of the patient.”5 At its most basic, phase-appropriate method development maps the “what is needed” to “when it is needed.”6 Effective phase-appropriate method development can provide long-term product support by introducing mass spectrometry compatibility and forced degradation development to ensure your methods are stability-indicating and amenable to unknown impurity identification. By instituting a phase-appropriate method development process, combined with a quality-by-design approacharound each logical sequence of events – and rigorously following it – it is more likely to create a cost-effective, successful outcome as you take the drug product through the regulatory process.
It Can Pay to Outsource
As the incentives for strong phase-appropriate method development increase, so too has the recognition of its value. Unfortunately, “it is not uncommon…to find pharmaceutical companies and contract research organizations (CROs) that are not taking advantage of the phase-appropriate approach and simply reference the typical ICH guidance for analytical items, such as method validation.”7 However, while FDA guidance encourages the use of a phase-appropriate approach, it is lacking in details and requirements. This leaves many companies to seek out ICH guidance as an alternative, conservative approach. Also, within their CGMP quality system, they may find it difficult to accommodate differing levels of CGMP compliance throughout the various clinical phases of development. This is when it might be an opportune moment to consider an outside expert that specializes in phase-appropriate method development processes for drug discovery and validation. A successful yet robust phase-appropriate method development program can balance competing interests and requirements and still provide a regimen that meets the overall development goals without sacrificing any of the requirements of the program.
AMPAC Analytical Laboratories (AAL), an SK pharmteco company, has decades of experience in providing a wide array of release testing services for raw materials, intermediates, APIs, and drug products. Our labs are equipped to handle hazardous, cytotoxic/high potency compounds as well as controlled substances for schedule II through V. Additionally, we have not only the expertise to conduct forced degradation experimentsbut also appropriate instrumentation like mass spectrometers to support later phases of development for your products. Please contact us to discuss how we can ensure the success of your drug discovery and development project and simultaneously reduce risks.
Forced Degradation is an important addendum to our previous post on Stability and Storage. Stressors are applied to new APIs and drug products to determine their degradation pathways and products under a variety of environmental conditions, including acid, base, light, heat, and oxidation. Forced degradation studies are also known as stress testing, stress studies, stress decomposition studies, and forced decomposition studies. These conditions “…are more severe than accelerated (stability) conditions and thus generate degradation products that can be studied to determine the stability of the molecule.”1
Regulatory requirements for forced degradation were enacted by the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) in 1993.2 However, these guidelines “are very general in (the) conduct of forced degradation and do not provide details about the practical approach towards stress testing. Although forced degradation studies are a regulatory requirement and scientific necessity during drug development, it is not considered as a requirement for (a) formal stability program.”1 However, stability studies have become a requisite for new drug moieties. In the absence of specific guidelines, the amount of stress needs to be representational: “Overstressing a molecule can lead to degradation profiles that are not representative of real storage conditions and perhaps not relevant to method development. Therefore, stress-testing conditions should be realistic and not excessive.”3
AMPAC Analytical (AAL), an SK pharmteco company, can assist with forced degradation studies for products at all phases of development, in tandem with stability, storage, and method development, to ensure the viability of the drug products as they were designed. We introduce forced degradation studies early in method development to ensure your product quality throughout the development lifecycle. Contact AAL today to learn more.








 The path to successful drug discovery and development is extremely long, expensive, and risky and can take between 10 to 15 years at an average cost of more than $1–2 billion for every new drug that is approved for clinical use.
The path to successful drug discovery and development is extremely long, expensive, and risky and can take between 10 to 15 years at an average cost of more than $1–2 billion for every new drug that is approved for clinical use.
 Forced Degradation is an important addendum to our
Forced Degradation is an important addendum to our