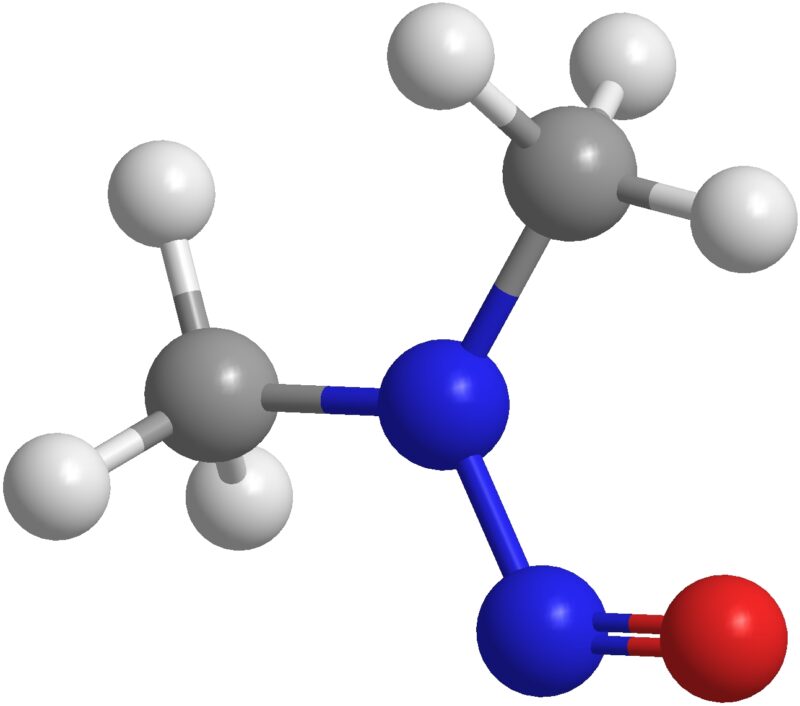
Forced Degradation is the testing of a drug product or drug substance using situations more taxing than those if conditions were simply accelerated. This type of stability testing is designed to demonstrate various degradation pathways within the product or substance and assists with the development of the product itself, along with its packaging. Within forced degradation testing, there is a subset of studies known as photo-stability. The guidelines are outlined by the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) and the FDA. As the ICH Topic Q1B document states, “The intrinsic photostability characteristics of new drug substances and products should be evaluated to demonstrate that, as appropriate, light exposure does not result in unacceptable change.”1
They recommend a range of different studies that address potential areas of concern:
A systematic approach to photostability testing is recommended covering, as appropriate, studies such as:
(i) Tests on the drug substance; (ii) Tests on the exposed drug product outside of the immediate pack; and, if necessary,
(iii) Tests on the drug product in the immediate pack; and, if necessary,
(iv) Tests on the drug product in the marketing pack.1
The type of lights used in photo-stability are also described. They offer options, including:
…any light source that is designed to produce an output similar to the D65/ID65 emission standard…
A cool white fluorescent lamp designed to produce an output similar to that specified in ISO 10977 (1993) …
A near UV fluorescent lamp having a spectral distribution from 320 nm to 400 nm with a maximum energy emission between 350 nm and 370 nm…1
Additionally, temperature control should be maintained to minimize changes and variations.
Finally, these guidelines also provide and elucidate the differences between procedures for drug products and drug substances. It is a crucial reference for administering or simply learning more about photo-stability forced degradation testing.
AMPAC Analytical has the equipment and expertise to provide comprehensive photo-stability studies for your products. Additionally, we have high-resolution and triple-quadrupole mass spectrometers to aid in the identification of potential degradants. Contact us today to learn how AMPAC Analytical can support your photo-stability needs.
(Per- and) PolyFluoroAlkyl Substances (PFAS) are a class of ubiquitous chemicals that have been found in water, air, fish, and soil across the nation and worldwide. Known as “Forever Chemicals,” there are thousands of different PFAS, and they are present in consumer, commercial, and industrial products.1 Having one of the strongest bonds in organic chemistry, their structures proved to be resistant to heat, water, oil, and degradation.2 They are found in “food packaging and non-stick cookware, cosmetics, waterproof and stain-proof textiles and carpet, aqueous film forming foam (AFFF) to fight Class B fires, and as part of metal plating processes.”3 Teflon and Scotchgard were two of the pioneering products to utilize these fluoropolymers. The two most common PFAS are perfluorooctanoic acid (PFOA) and perfluorooctane sulfonic acid (PFOS).
Health Concerns of PFAS
Some of the most frequently cited health concerns associated with PFAS include adverse cardiovascular, immunity, developmental, and hepatic effects.3,4The most commonly heard refrain to minimize these concerns is that if they are so prevalent, why are there
not more health issues associated with them? In fact, “The Lancet Commission on Pollution and Health reported that pollution was responsible for 9 million premature deaths in 2015, making it the world’s largest environmental risk factor for disease and
premature death.” This was updated in 2019, and those numbers held steady, accounting for one in six deaths worldwide.5/i> While this number includes all types of pollution, the impacts are clear.
The Exposure Concerns of PFAS Are Regulatory and Legal
Due to their combination of persistence, pervasiveness, mobility, and the ability of some to bioaccumulate (or build up in animals and humans), they have been in the news recently too.6 Predictably, they are also now moving through the courts.7-9 Some of the most common areas of litigation are directed at PFAS found in drinking water and firefighting foam. The regulatory initiatives are also increasing. These address a range from water and soil to numerous manmade products including food packaging.10,11 The European Chemicals Agency (ECHA) and the NIH have a wealth of guidance and regulations that apply to PFAS.12,13
PFAS Detection
The EPA has useful direction for analytical methods development and sampling research that outlines the “laboratory validation process following a particular rulemaking or guidance effort and are available to support regulatory or guidance activities.”14 For
technique and equipment, PFAS are typically analyzed by mass spectrometry, coupled with gas chromatography or liquid chromatography (GCMS and LCMS), which enables detection in the low parts per billion.
AMPAC Analytical has years of experience and numerous experts in trace analysis by mass spectrometry who can assist with method development for high-volume analyses for both common and atypical sample matrices that will allow you to stay ahead of evolving regulatory concerns. Please contact us with specific questions or to receive a quote for PFAS quantitation.
The linkage between nitrosamines and cancer was first postulated by William Lijinsky in 1970. Then, in 2018, N-nitroso-dimethylamine (NDMA)) was detected in an active pharmaceutical ingredient, Valsartan (an Angiotensin-II-receptor antagonist). Finally, the FDA issued a guidance for the industry, “Control of Nitrosamine Impurities in Human Drugs”, in the fall of 2020. However, the guidelines continue to evolve. There has been an update in March of 2021, with ongoing risk assessments. Other regulatory agencies have instituted their own, along with updates. For example, since our blog series on nitrosamines, there have been some regulatory updates from the European Medicines Agency (EMA). Their new guidelines are outlined in a document entitled, “Questions and Answers for Marketing Authorization Holders/Applicants on the CHMP Opinion for the Article 5(3) of Regulation (EC) No 726/2004 Referral on Nitrosamine Impurities in Human Medicinal Products.”1 The updated section answers the crucial question, “Which limits apply for nitrosamines in medicinal products?”
The answers are provided with a definition of nitrosamines and acceptable exposures:
“The ICH M7(R1) guideline defines N-nitrosamines as substances of the “cohort of concern” for which limits in medicinal products refer to the so-called substance-specific acceptable intake (AI) (the Threshold of Toxicological Concern, TTC, value of 1.5 ug/day cannot be routinely applied) which is associated with a negligible risk (theoretical excess cancer risk of <1 in 100,000 over a lifetime of exposure). The calculation of AI assumes a lifelong daily administration of the maximum daily dose of the medicinal product and is based on the approach outlined in the ICH M7(R1) guideline as well as the principles described in relation to the toxicological evaluation in the assessment report of the CHMP’s Article 5(3) opinion on nitrosamine impurities in human medicinal products.”1,2 (A previous blog examined TTC, here.)
A Useful List of Nitrosamine Limits
In Appendix 1 found on the EMA site, there is a list of more than eighty nitrosamines listed, along with CAS numbers, known medicinal sources, CPCA (Carcinogenic Potency Categorization Approach) categories, and their guidance publication dates.3,4
Some Caveats
There are some exceptions that should be considered. For example, the EMA states, “The ‘less than lifetime’ (LTL) approach should not be applied in calculating the limits as described above but can only be considered after consultation with competent authorities as a temporary measure until further measures can be implemented to reduce the contaminant at or below the limits defined above.”1
Additionally, those medications intended for advanced cancers also have some exceptions. For example, “If the active substance itself is mutagenic or clastogenic at therapeutic concentrations, N-nitrosamine impurities should be controlled at limits for non-mutagenic impurities according to ICH M7(R1).”
There is also guidance when one or more than one nitrosamines may be present. For the latter, the guidance advises one of two approaches:
The total daily intake of all identified N-nitrosamines is not to exceed the AI of the most potent N-nitrosamine identified.
The total risk level calculated for all identified N-nitrosamines is not to exceed 1 in 100,000. The approach chosen needs to be duly justified by the MAH (Marketing Authorization Holder)/Applicant.1
Final Thoughts on Nitrosamines
Nitrosamine guidance worldwide is ever-evolving, yet the impetus to quantify and regulate them is clear. There will doubtlessly be further updates to regulations. AMPAC Analytical Laboratories – an SK pharmteco company (AAL) is an industry leader in the detection of nitrosamines and other genotoxic impurities (GTI), We have the specialized expertise, equipment, and methodologies to detect these impurities by gas chromatography or high-performance liquid chromatography coupled with mass spectrometry to support your API project. Also, importantly, AAL can assist in navigating those projects within today’s regulatory landscape. Please contact us with any specific questions or to receive a quote for nitrosamines or other GTIs.
Extractables and Leachables (E&L) are essential areas of concern for the pharmaceutical and food industries, specifically regarding their packaging, usage components (e.g., medical devices or syringes), and the manufacturing chain. We will examine testing of analysis of them within pharmaceutical applications. The two terms are related but distinct, each with its own analytical requirements.
Definitions of Extractables and Leachables
A handy article published in Pharmaceutical Engineering by the International Society for Pharmaceutical Engineering (ISPE) explains that “Extractables are chemical compounds that migrate from single-use systems (SUS) into model solvent solutions under controlled and exaggerated conditions depending on temperature, pH, polarity, and time.” In other words, this happens when using strong solvents. They note that “SUS are normally not exposed to such conditions in biopharmaceutical processes.”1
ISPE’s article defines leachables as “chemical compounds that migrate from SUS into process solutions under normal biopharmaceutical process conditions. They further clarify that these compounds “may end up in the final drug product formulation. For the most part, leachables are a subset of extractables, although interaction with product components may produce leachables not seen as extractables.”1
Guidance on Extractables and Leachables
The FDA has released a series of guidelines for the pharmaceutical industry, including Container Closure Systems for Packaging Human Drugs and Biologics, that provide guidance for submission in support of an original application for any drug product. It also covers a wide range of forms and delivery systems of drugs:
Inhaled
Injected
Liquid-based
Oral
Solid oral dosage forms
Ophthalmic
Topical and topical delivery systems
Powders for reconstitution
And other dosage forms
Additionally, the International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH) also has issued the ICH Q3E: Guideline for Extractables and Leachables.2,3 These are both useful in providing direction for E&L concerns and control strategies.
Plan Against Extractables and Leachables
To guarantee adherence to all guidelines and regulations while ensuring patient safety, it is crucial to know and utilize materials compatible with your product. To accomplish this, solvent use, packaging, and delivery systems must all be tested and analyzed in cGMP and FDA-compliant laboratories. This should include the following:
A thorough review of all materials used in packaging and production, production,and equipment to predict the compatibility of your packaging system with your product. AAL can provide reports for items from each step.
Extraction studies on the materials used.
Leachable studies to identify any impurity resulting from those materials found in the final product under normal usage conditions.
If impurities are detected, AAL can provide toxicological evaluations, including profiles of the impurities and the risks they pose for the patients, establish safety limits, or adjust for different forms of medication application.
We can assess risks created by various exposure levels due to the impurity in the finished product.
Finally, AAL provides a detailed report of our findings in accordance with the applicable governing bodies (e.g., FDA, EMA, PQRI, PDA).
AMPAC Analytical can review your analysis and testing needs for extractables and leachables for any forms and delivery systems listed in the table above, complying with the strictest standards necessary.




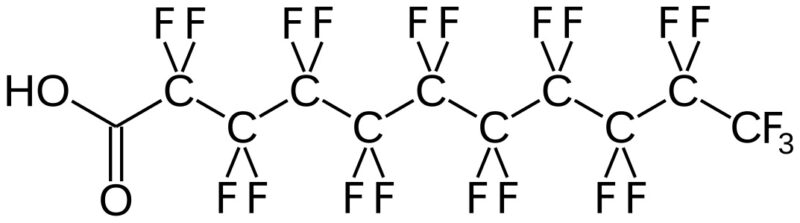
 (Per- and) PolyFluoroAlkyl Substances (PFAS) are a class of ubiquitous chemicals that have been found in water, air, fish, and soil across the nation and worldwide. Known as “Forever Chemicals,” there are thousands of different PFAS, and they are present in consumer, commercial, and industrial products.1 Having one of the strongest bonds in organic chemistry, their structures proved to be resistant to heat, water, oil, and degradation.2 They are found in “food packaging and non-stick cookware, cosmetics, waterproof and stain-proof textiles and carpet, aqueous film forming foam (AFFF) to fight Class B fires, and as part of metal plating processes.”3 Teflon and Scotchgard were two of the pioneering products to utilize these fluoropolymers. The two most common PFAS are perfluorooctanoic acid (PFOA) and perfluorooctane sulfonic acid (PFOS).
(Per- and) PolyFluoroAlkyl Substances (PFAS) are a class of ubiquitous chemicals that have been found in water, air, fish, and soil across the nation and worldwide. Known as “Forever Chemicals,” there are thousands of different PFAS, and they are present in consumer, commercial, and industrial products.1 Having one of the strongest bonds in organic chemistry, their structures proved to be resistant to heat, water, oil, and degradation.2 They are found in “food packaging and non-stick cookware, cosmetics, waterproof and stain-proof textiles and carpet, aqueous film forming foam (AFFF) to fight Class B fires, and as part of metal plating processes.”3 Teflon and Scotchgard were two of the pioneering products to utilize these fluoropolymers. The two most common PFAS are perfluorooctanoic acid (PFOA) and perfluorooctane sulfonic acid (PFOS).